Alpha-L-iduronate deficiency; Mucopolysaccharidosis type I; Severe MPS I; Attenuated MPS I; MPS I H; MPS I S; Hurler syndrome; Scheie syndrome; Hurler-Scheie syndrome; MPS 1 H/S; Lysosomal storage disease - mucopolysaccharidosis type I DefinitionMucopolysaccharidosis type I (MPS I) is a rare disease in which the body is missing or does not have enough of an enzyme needed to break down long chains of sugar molecules. These chains of molecules are called glycosaminoglycans (formerly called mucopolysaccharides). As a result, the molecules build up in different parts of the body and cause various health problems. The condition belongs to a group of diseases called mucopolysaccharidoses (MPSs). MPS I is the most common. There are several other types of MPSs, including: CausesMPS I is inherited, which means that your parents typically pass the disease on to you. If both parents carry a nonworking copy of the gene related to this condition, each of their children has a 25% (1 in 4) chance of developing the disease. People with MPS I do not make an enzyme called lysosomal alpha-L-iduronidase. This enzyme helps break down long chains of sugar molecules called glycosaminoglycans. These molecules are found throughout the body, often in mucus and in fluid around the joints. Without the enzyme, glycosaminoglycans build up and damage organs, including the heart. Symptoms can range from mild to severe. The mild form is called attenuated MPS I and the severe form is called severe MPS I. SymptomsSymptoms of MPS I most often appear from ages 3 to 8 years. Children with severe MPS I develop symptoms earlier than those with the less severe form. Some of the symptoms include:
Exams and TestsIn some states, babies are tested for MPS I as part of the newborn screening tests. Other tests that may be done depending on symptoms include:
TreatmentEnzyme replacement therapy may be recommended. The medicine, called laronidase (Aldurazyme), is given through a vein (IV, intravenously). It replaces the missing enzyme. Talk to your child's health care provider for more information. Bone marrow transplant has been tried. The treatment has had mixed results. Other treatments depend on the organs that are affected. Support GroupsMore information and support for people with MPS I and their families can be found at:
Outlook (Prognosis)Children with severe MPS I usually do not do well. Their health problems worsen over time, resulting in death by age 10. Children with attenuated (milder) MPS I have fewer health problems, with many leading fairly normal lives into adulthood. When to Contact a Medical ProfessionalContact your provider if:
PreventionExperts recommend genetic counseling and testing for couples with a family history of MPS I who are considering having children. Prenatal testing is available. ReferencesKumar V, Abbas AK, Aster JC. Genetic disorders. In: Kumar V, Abbas AK, Aster JC, eds. Robbins & Cotran Pathologic Basis of Disease. 10th ed. Philadelphia, PA: Elsevier; 2021:chap 5. Lampe C. Mucopolysaccharidoses. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 109. Pyeritz RE. Inherited diseases of connective tissue. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 239. Turnpenny PD, Ellard S, Cleaver R. Inborn errors of metabolism. In: Turnpenny PD, Ellard S, Cleaver R, eds. Emery's Elements of Medical Genetics and Genomics. 16th ed. Philadelphia, PA: Elsevier; 2022:chap 18. | ||
| ||
Review Date: 4/8/2025 Reviewed By: Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. View References The information provided herein should not be used during any medical emergency or for the diagnosis or treatment of any medical condition. A licensed medical professional should be consulted for diagnosis and treatment of any and all medical conditions. Links to other sites are provided for information only -- they do not constitute endorsements of those other sites. No warranty of any kind, either expressed or implied, is made as to the accuracy, reliability, timeliness, or correctness of any translations made by a third-party service of the information provided herein into any other language. © 1997- A.D.A.M., a business unit of Ebix, Inc. Any duplication or distribution of the information contained herein is strictly prohibited. | ||
| ||


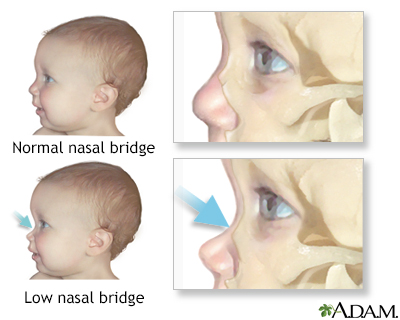 Low nasal bridge
Low nasal bridge