Testicular feminization; AIS - complete; AIS - partial; AIS - mild; Androgen insensitivity syndrome - differences of sex development; Androgen insensitivity syndrome - Intersex DefinitionAndrogen insensitivity syndrome (AIS) is when a person who has one X and one Y chromosome (typically seen in males) is resistant to hormones that produce a male appearance (called androgens). As a result, the person has some of the physical traits of a female, but the genetic makeup of a male. Androgen insensitivity syndrome is one of the conditions that are described as differences of sex development (DSD). CausesIn the first 2 to 3 months of pregnancy, all babies have the same genitals. As a baby grows inside the womb, male or female genitals develop depending on the sex chromosomes the baby has from the parents (XY for male, XX for female). It also depends on the levels of androgens. In a baby with XY chromosomes, high levels of androgens are made in the testes. This baby will develop male genitals. In a baby with XX chromosomes, there are no testes and the levels of androgens are very low. This baby will develop female genitals. AIS is caused by genetic defects on the X chromosome. These defects make the body unable to respond to the hormones that produce a male appearance. The syndrome is divided into three main categories:
In CAIS:
In PAIS:
In MAIS:
The syndrome is passed down genetically (X-linked recessive inheritance). People with two X chromosomes are not affected if only one copy of the X chromosome carries the genetic variant. Males who inherit the gene from their mothers will have the condition. There is a 50% chance that a male child of a mother with the genetic trait will be affected. Every female child of a mother with the genetic trait has a 50% chance of carrying the genetic trait. Family history is important in determining risk factors of AIS. SymptomsA person with CAIS appears to be female but has no uterus, no fallopian tubes, or ovaries. They have very little armpit and pubic hair. At puberty, female sex characteristics (such as breasts) develop. However, the person does not menstruate or become fertile. People with PAIS may have both male and female physical characteristics (ambiguous genitalia). These characteristics vary from person to person. These may include:
A person with MAIS has male genitals and may have:
Exams and TestsCAIS is rarely discovered during childhood. Sometimes, a growth is felt in the abdomen or groin that turns out to be a testicle when it is explored with surgery. Most people with this condition are not diagnosed until they do not get a menstrual period. PAIS is often discovered during childhood because the person may have both male and female physical traits. Tests used to diagnose this condition may include:
Other blood tests may be done to help tell the difference between AIS and androgen deficiency. TreatmentIn children with CAIS, testicles that are in the wrong place may not be removed until a child finishes growing and goes through puberty. At this time, the testes may be removed because they can develop cancer, just like any undescended testicle. Estrogen replacement may be prescribed after puberty. Females with CAIS have a shortened vagina. They may choose to use vaginal dilation to lengthen it. Infants with PAIS may be assigned a gender depending on the extent of genital ambiguity. However, gender assignment is a complex issue, and the need for it and the timing of it must be considered carefully. PAIS can be distressing for parents and families. While early surgery may make the parents feel more comfortable, the child may not be happy with the decision as they become older. Many health experts and intersex advocates suggest waiting until the child is old enough to be involved in the decision, unless surgery is needed for the health of the infant. Possible treatments for PAIS include:
Treatment and gender assignment can be a very complex issue and must be targeted to each individual person. Treatment guidelines are still evolving. It is vital that children with AIS and their parents receive care and support from a health care team that includes different specialists with expertise in gender medicine. This should include mental health professionals to help provide support for both children and their parents. Support GroupsMore information and support for people with AIS and their families can be found at:
Outlook (Prognosis)Androgens are most important during early development in the womb. People with AIS can have a normal lifespan and be totally healthy, but they may have difficulty conceiving a child. Possible ComplicationsComplications include:
When to Contact a Medical ProfessionalContact your health care provider if you or your child has signs or symptoms of the syndrome. Genetic testing and counseling are recommended if AIS is suspected. ReferencesChan Y-M, Hannema SE, Achermann JC, Hughes IA. Disorders of sex development. In: Melmed S, Auchus, RJ, Goldfine AB, Koenig RJ, Rosen CJ, eds. Williams Textbook of Endocrinology. 14th ed. Philadelphia, PA: Elsevier; 2020:chap 24. Délot EC, Vilain E. Differences of sex development. In: Strauss JF, Barbieri R, Dokras A, Williams CJ, Williams Z, eds. Yen & Jaffe's Reproductive Endocrinology. 9th ed. Elsevier; 2024:chap 17. Donohoue PA. Disorders of sex development. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 606. Genetic and Rare Diseases Information Center website. Androgen insensitivity syndrome. rarediseases.info.nih.gov/diseases/5803/androgen-insensitivity-syndrome. Updated February 2024. Accessed March 20, 2024. Matsumoto AM, Anawalt BD. Testicular disorders. In: Melmed S, Auchus RJ, Goldfine AB, Koenig RJ, Rosen CJ, eds. Williams Textbook of Endocrinology. 14th ed. Philadelphia, PA: Elsevier; 2020:chap 19. Shnorhavorian M, Fechner PY. Differences in sex development. In: Gleason CA, Sawyer T, eds. Avery's Diseases of the Newborn. 11th ed. Philadelphia, PA: Elsevier; 2024:chap 85. Yu RN, Diamond DA. Disorders of sexual development: etiology, evaluation, and medical management. In: Partin AW, Dmochowski RR, Kavoussi LR, Peters CA, eds. Campbell-Walsh-Wein Urology. 12th ed. Philadelphia, PA: Elsevier; 2021:chap 48. | ||
| ||
Review Date: 3/12/2024 Reviewed By: Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. View References The information provided herein should not be used during any medical emergency or for the diagnosis or treatment of any medical condition. A licensed medical professional should be consulted for diagnosis and treatment of any and all medical conditions. Links to other sites are provided for information only -- they do not constitute endorsements of those other sites. No warranty of any kind, either expressed or implied, is made as to the accuracy, reliability, timeliness, or correctness of any translations made by a third-party service of the information provided herein into any other language. © 1997- A.D.A.M., a business unit of Ebix, Inc. Any duplication or distribution of the information contained herein is strictly prohibited. | ||
| ||


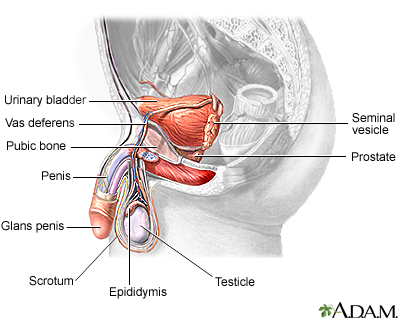 Male reproductive ...
Male reproductive ...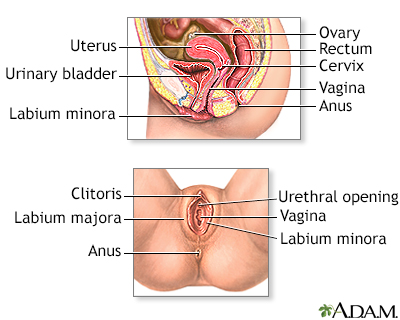 Female reproductiv...
Female reproductiv...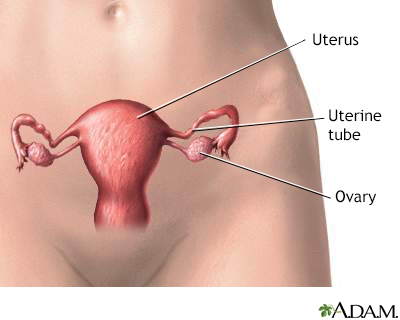 Female reproductiv...
Female reproductiv... Karyotyping
Karyotyping