FCU; Fenilcetonuria neonatal DefiniciónEs una afección poco frecuente en la cual un bebé nace sin la capacidad para descomponer apropiadamente un aminoácido llamado fenilalanina. CausasLa fenilcetonuria es una enfermedad hereditaria. Si ambos padres portan una copia del gen disfuncional relacionado con esta afección, cada uno de sus hijos tiene un 24% (1 de 4) de riesgo de desarrollar la enfermedad. Esto se denomina herencia autosómica recesiva. Los bebés con fenilcetonuria carecen de una enzima denominada fenilalanina hidroxilasa. Esta necesaria para descomponer el aminoácido esencial fenilalanina. La fenilalanina se encuentra en alimentos que contienen proteína. Sin la enzima, los niveles de fenilalanina se acumulan en el cuerpo. Esta acumulación puede dañar el sistema nervioso central y ocasionan daño cerebral. SíntomasLa fenilalanina juega un papel en la producción corporal de melanina. El pigmento es responsable de dar color a la piel y al cabello. Por lo tanto, los niños con esta afección usualmente tienen un cutis, cabello y ojos más claros que sus hermanos o hermanas sin la enfermedad. Otros síntomas pueden incluir:
Si la fenilcetonuria se deja sin tratamiento o si se consumen alimentos que contienen fenilalanina, el aliento, la piel, el cerumen y la orina pueden tener un olor "a ratón" o "a moho". Este olor se debe a la acumulación de sustancias de fenilalanina en el cuerpo. Pruebas y exámenesLa fenilcetonuria se puede detectar fácilmente con un simple examen de sangre. Todos los estados de los Estados Unidos exigen una prueba de detección de esta enfermedad para todos los recién nacidos, como parte del grupo de pruebas de detección para neonatos. Este examen generalmente se lleva a cabo tomando unas cuantas gotas de sangre del bebé antes de que este salga del hospital. Si la prueba de detección es positiva, se requieren exámenes adicionales de sangre y orina para confirmar el diagnóstico. También se llevan a cabo pruebas genéticas. TratamientoLa fenilcetonuria es una enfermedad que se puede tratar. El tratamiento comprende una dieta muy baja en fenilalanina, especialmente cuando el niño está creciendo. La dieta se tiene que seguir en forma estricta. Esto requiere la supervisión exhaustiva por parte del dietista certificado o del proveedor de atención médica y la cooperación de los padres y del niño. Quienes que continúan con la dieta hasta la vida adulta tienen una mejor salud física y mental que quienes no lo hacen. Una “dieta de por vida” se ha convertido en la pauta que la mayoría de los expertos recomiendan. Las mujeres que presentan fenilcetonuria deben seguir la dieta antes de la concepción y durante todo el embarazo. Hay grandes cantidades de fenilalanina en la leche, los huevos y otros alimentos comunes. El edulcorante artificial Nutrasweet (aspartamo) también contiene fenilalanina. Cualquier producto que contenga aspartamo se debe evitar. Hay muchas leches en polvo especiales hechas para bebés con fenilcetonuria. Los bebés con fenilcetonuria pueden alimentarse con leche materna de manera segura, junto con el uso de alimentos médicos y un control adecuado de los niveles de fenilalanina. Estos alimentos médicos especiales se pueden usar como una fuente de proteína, con un contenido extremadamente bajo en fenilalanina y balanceada con respecto a los aminoácidos esenciales restantes. Los niños mayores y los adultos usan una leche en polvo distinta que proporciona proteína en las cantidades que ellos necesitan. Las personas con fenilcetonuria necesitan tomar la leche en polvo todos los días por el resto de su vida. Expectativas (pronóstico)Se espera que el desenlace clínico sea muy alentador si la dieta se sigue estrictamente, comenzando poco después del nacimiento del niño. Si el tratamiento se retrasa o el trastorno permanece sin tratamiento, se presentará daño cerebral. El desempeño escolar se puede deteriorar levemente. Si no se evitan las proteínas que contengan fenilalanina, la fenilcetonuria puede conducir a discapacidad mental hacia el final del primer año de vida. Posibles complicacionesSi este trastorno no recibe tratamiento, se presenta discapacidad mental grave. El trastorno de hiperactividad y déficit de atención (THDA) o ADHD, por sus siglas en inglés, parece ser un problema común en quienes no siguen estrictamente una dieta muy baja en fenilalanina. Cuándo contactar a un profesional médicoComuníquese con su proveedor si a su bebé no le han realizado exámenes para fenilcetonuria. Esto es particularmente importante si algún familiar tiene este trastorno. PrevenciónUn análisis enzimático o una prueba genética pueden determinar si los padres son portadores del gen de la fenilcetonuria (FCU). Asimismo, se puede tomar una muestra de vellosidades coriónicas o amniocentesis durante el embarazo para examinar el feto en búsqueda de esta enfermedad. Es muy importante que las mujeres con fenilcetonuria sigan estrictamente una dieta baja en fenilalanina, tanto antes de quedar embarazadas como a través de todo el embarazo. La acumulación de la fenilalanina le causará daño al bebé en desarrollo, incluso si el niño no heredó la enfermedad completa. ReferenciasDietzen DJ, Willrich MAV. Amino acids, peptides, and proteins. In: Rifai N, Chiu RWK, Young I, Burnham C-A D, Wittwer CT, eds. Tietz Textbook of Laboratory Medicine. 7th ed. St Louis, MO: Elsevier; 2023:chap 31. Kliegman RM, St. Geme JW, Blum NJ, et al. Defects in metabolism of amino acids. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 105. Kumar V, Abbas AK, Aster JC, Deyrup AT, Das A. Genetic and pediatric diseases. In: Kumar V, Abbas AK, Aster JC, Deyrup AT, Das A, eds. Robbins & Kumar Basic Pathology. 11th ed. Philadelphia, PA: Elsevier; 2023:chap 4. Merritt JL, Gallagher RC. Inborn errors of carbohydrate, ammonia, amino acid, and organic acid metabolism. In: Gleason CA, Sawyer T, eds. Avery's Diseases of the Newborn. 11th ed. Philadelphia, PA: Elsevier; 2024:chap 29. Smith WE, Berry SA, Bloom K, et al. Phenylalanine hydroxylase deficiency diagnosis and management: A 2023 evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2025;27(1):101289. PMID: 39630157 pubmed.ncbi.nlm.nih.gov/39630157/. | ||
| ||
Actualizado : 4/8/2025 Versión en inglés revisada por : Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. Ver referencias La información aquí contenida no debe utilizarse durante ninguna emergencia médica, ni para el diagnóstico o tratamiento de alguna condición médica. Debe consultarse a un médico con licencia para el diagnóstico y tratamiento de todas y cada una de las condiciones médicas. Los enlaces a otros sitios se proporcionan sólo con fines de información, no significa que se les apruebe. No se otorga garantía de ninguna clase, ya sea expresa o implícita, en cuanto a la precisión, confiabilidad, actualidad o exactitud de ninguna de las traducciones hechas por un proveedor de servicios externo de la información aquí contenida en otro idioma. © 1997- A.D.A.M., unidad de negocios de Ebix, Inc. La reproducción o distribución parcial o total de la información aquí contenida está terminantemente prohibida. | ||
| ||


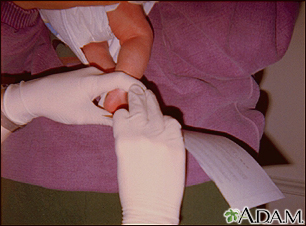 Examen de fenilceto...
Examen de fenilceto...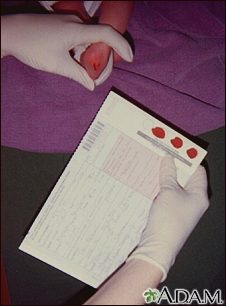 Prueba de tamizaje ...
Prueba de tamizaje ...