Enfermedad de Werdnig-Hoffmann; Enfermedad de Kugelberg-Welander DefiniciónLa atrofia muscular espinal (AME) es un grupo de trastornos de las neuronas motoras (células motoras). Estos trastornos se transmiten de padres a hijos (hereditarios) y pueden aparecer en cualquier etapa de la vida. El trastorno lleva a debilidad y atrofia muscular. CausasLa AME es un grupo de diferentes enfermedades de los nervios motores (o neuronas). La enfermedad es causada por una falta de proteína (SMN) debido a genes defectuosos. La mayoría de las veces, una persona tiene que heredar una copia del gen defectuoso de ambos padres para resultar afectada. La forma más grave es la AME tipo I, también llamada enfermedad de Werdnig-Hoffman. Los bebés con AME tipo II tienen síntomas menos intensos a comienzos del período de lactancia, pero se debilitan con el tiempo. La AME tipo III es una forma menos grave de la enfermedad. En casos poco frecuentes, la AME comienza en la adultez. Esta es la forma más leve de la enfermedad. Un antecedente familiar de AME en un familiar inmediato (como hermano o hermana) es un factor de riesgo para todos los tipos de este trastorno. SíntomasLos síntomas de la AME pueden variar dependiendo de su tipo:
A menudo, la debilidad se nota primero en los músculos de los hombros y las piernas. La debilidad empeora con el tiempo y finalmente se vuelve intensa. Síntomas en un bebé:
Síntomas en un niño:
Con la AME, los nervios que controlan la sensación (nervios sensoriales) no se afectan. Por lo tanto, una persona con esta enfermedad puede sentir todo normalmente. Pruebas y exámenesEl proveedor de atención médica elaborará una historia clínica cuidadosa y llevará a cabo un examen del sistema nervioso y del cerebro (neurológico) para averiguar si hay:
Los exámenes que se pueden ordenar incluyen:
TratamientoNo hay ningún tratamiento para curar la AME. Sin embargo, ahora hay tres fármacos aprobados por la FDA que disminuyen la velocidad en la que progresa la debilidad muscular:
Estos fármacos ayudan al aumentar la cantidad de la proteína SMN producida. Converse con su proveedor para ver si alguno de estos medicamentos es el correcto para usted o su hijo. Los cuidados complementarios son importantes. Las complicaciones respiratorias son comunes en las formas más graves de la AME. Para ayudar con la respiración, se puede necesitar un dispositivo o máquina llamado respirador artificial. También se debe vigilar a las personas con AME para evitar el ahogamiento. Esto es porque los músculos que controlan la deglución son débiles. La fisioterapia es importante para prevenir contracciones de los músculos y tendones, y la curvatura anormal de la columna (escoliosis). Puede ser necesario el uso de dispositivos ortopédicos. Se puede necesitar cirugía para corregir las deformidades esqueléticas, como la escoliosis. Expectativas (pronóstico)Los niños con la AME tipo I rara vez viven por más de 2 a 3 años, debido a los problemas respiratorios y las infecciones. El tiempo de supervivencia con el tipo II es mayor, pero la enfermedad mata a la mayoría de las personas afectadas cuando todavía son niños. Los niños con el tipo III de la enfermedad pueden sobrevivir hasta comienzos de la edad adulta. Sin embargo, las personas con todas las formas de la enfermedad sufren de debilidad y fatiga que empeoran con el tiempo. Los adultos que desarrollan atrofia muscular espinal a menudo tienen una expectativa normal. Posibles complicacionesLas complicaciones que pueden presentarse a raíz de la AME incluyen:
Cuándo contactar a un profesional médicoComuníquese con su proveedor si su hijo:
La dificultad respiratoria puede convertirse rápidamente en una situación de emergencia. PrevenciónSe recomienda la asesoría genética para las personas con antecedentes familiares de AME que deseen tener hijos. ReferenciasFearon C, Murray B, Mitsumoto H. Disorders of upper and lower motor neurons. In: Jankovic J, Mazziotta JC, Pomeroy SL, Newman NJ, eds. Bradley and Daroff's Neurology in Clinical Practice. 8th ed. Philadelphia, PA: Elsevier; 2022:chap 97. Manzur AY. Evaluation and investigation of neuromuscular disorders. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 647. NIH-National Institute of Neurological Disorders and Stroke website. Spinal muscular atrophy. www.ninds.nih.gov/health-information/disorders/spinal-muscular-atrophy. Updated November 28, 2023. Accessed March 14, 2024. | ||
| ||
Actualizado : 12/31/2023 Versión en inglés revisada por : Joseph V. Campellone, MD, Department of Neurology, Cooper Medical School at Rowan University, Camden, NJ. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. Ver referencias La información aquí contenida no debe utilizarse durante ninguna emergencia médica, ni para el diagnóstico o tratamiento de alguna condición médica. Debe consultarse a un médico con licencia para el diagnóstico y tratamiento de todas y cada una de las condiciones médicas. Los enlaces a otros sitios se proporcionan sólo con fines de información, no significa que se les apruebe. No se otorga garantía de ninguna clase, ya sea expresa o implícita, en cuanto a la precisión, confiabilidad, actualidad o exactitud de ninguna de las traducciones hechas por un proveedor de servicios externo de la información aquí contenida en otro idioma. © 1997- A.D.A.M., unidad de negocios de Ebix, Inc. La reproducción o distribución parcial o total de la información aquí contenida está terminantemente prohibida. | ||
| ||


 Músculos superficia...
Músculos superficia...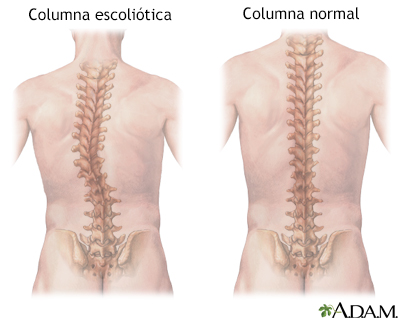 Escoliosis
Escoliosis