Degeneración hepatolenticular DefiniciónEs un trastorno hereditario que causa que haya demasiado cobre en los tejidos del cuerpo. El exceso de cobre causa daño al hígado y al sistema nervioso. CausasLa enfermedad de Wilson es un trastorno hereditario poco común. En caso de que ambos padres porten un gen disfuncional (variante) para la enfermedad de Wilson, hay un 25% de probabilidades en cada embarazo de que el niño tenga el trastorno. La enfermedad de Wilson hace que el cuerpo absorba y conserve demasiado cobre. Este cobre se deposita en el hígado, el cerebro, los riñones y los ojos. Esto ocasiona daño y muerte tisular y cicatrización. Los órganos afectados dejen de funcionar normalmente. Esta enfermedad es más común en personas de Europa oriental, Sicilia y el sur de Italia, aunque se puede presentar en cualquier grupo. Los síntomas de la enfermedad de Wilson aparecen típicamente en personas menores de 40 años. El daño al hígado empieza en los niños a la edad de 6 años, pero los síntomas clínicos con frecuencia se presentan en las personas en su adolescencia o a principios de los veinte. SíntomasLos síntomas pueden incluir:
Pruebas y exámenesUn examen de los ojos con lámpara de hendidura puede mostrar:
Un examen físico puede mostrar señales de:
Los exámenes de laboratorio pueden incluir:
Si hay problemas con el hígado, los exámenes de laboratorio pueden encontrar: Otros exámenes pueden incluir:
Las copias disfuncionales del gen llamado ATP7B causa la enfermedad de Wilson. Están disponibles pruebas de ADN para este gen. Hable con su proveedor de atención médica o con un asesor genético para saber si debe hacerse pruebas para detectar este gen. TratamientoEl objetivo del tratamiento es reducir la cantidad de cobre en los tejidos. Esto se hace mediante un procedimiento llamado quelación. Se proporcionan ciertos medicamentos que se fijan al cobre y ayudan a eliminarlo a través de los riñones o los intestinos. El tratamiento debe hacerse de por vida. Se pueden utilizar los siguientes medicamentos:
También se pueden utilizar los suplementos de vitamina E. Algunas veces, los medicamentos que quelan el cobre (como la penicilamina) pueden afectar el funcionamiento del cerebro y del sistema nervioso (función neurológica). Existen otros medicamentos bajo investigación que se fijan al cobre sin afectar la función neurológica. También se puede recomendar una dieta baja en cobre. Los alimentos que se deben evitar incluyen:
Es posible que prefiera beber agua destilada, porque en algunas partes el agua de grifo fluye a través de tubos de cobre. Evite el uso de utensilios de cocina hechos de cobre. Los síntomas se pueden tratar con ejercicios o fisioterapia. Las personas que se encuentran confundidas o que son incapaces de cuidar de sí mismas pueden necesitar medidas de protección especiales. En los casos en donde se presente daño hepático grave a causa de la enfermedad, se puede pensar en la posibilidad de un trasplante de hígado. Grupos de apoyoPuede encontrar más información y apoyo para las personas con la enfermedad de Wilson y sus familias en:
Expectativas (pronóstico)Se requiere tratamiento de por vida para controlar la enfermedad de Wilson. La enfermedad puede causar efectos mortales, tales como la pérdida de la función hepática. El cobre puede tener efectos tóxicos sobre el sistema nervioso. En los casos en los que la enfermedad no es mortal, los síntomas pueden ser incapacitantes. Posibles complicacionesLas complicaciones pueden incluir:
La insuficiencia hepática y el daño al sistema nervioso central (cerebro, médula espinal) son los efectos más comunes y peligrosos del trastorno. Si la afección no se detecta y se trata a tiempo, puede ser mortal. Cuándo contactar a un profesional médicoComuníquese con su proveedor si tiene síntomas de la enfermedad de Wilson. De igual forma, comuníquese con un asesor genético si tiene antecedentes de esta enfermedad en su familia y está planeando tener hijos. PrevenciónSe recomienda la asesoría genética para personas con antecedentes familiares o personales de enfermedad de Wilson. ReferenciasNational Institute of Diabetes and Digestive and Kidney Diseases website. Wilson disease. www.niddk.nih.gov/health-information/liver-disease/wilson-disease/definition-facts. Updated October 2018. Accessed September 13, 2024. Roberts EA. Wilson disease. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran's Gastrointestinal and Liver Disease. 11th ed. Philadelphia, PA: Elsevier; 2021:chap 76. Schilsky ML. Wilson disease. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 195. | ||
| ||
Actualizado : 8/18/2024 Versión en inglés revisada por : Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. Ver referencias La información aquí contenida no debe utilizarse durante ninguna emergencia médica, ni para el diagnóstico o tratamiento de alguna condición médica. Debe consultarse a un médico con licencia para el diagnóstico y tratamiento de todas y cada una de las condiciones médicas. Los enlaces a otros sitios se proporcionan sólo con fines de información, no significa que se les apruebe. No se otorga garantía de ninguna clase, ya sea expresa o implícita, en cuanto a la precisión, confiabilidad, actualidad o exactitud de ninguna de las traducciones hechas por un proveedor de servicios externo de la información aquí contenida en otro idioma. © 1997- A.D.A.M., unidad de negocios de Ebix, Inc. La reproducción o distribución parcial o total de la información aquí contenida está terminantemente prohibida. | ||
| ||


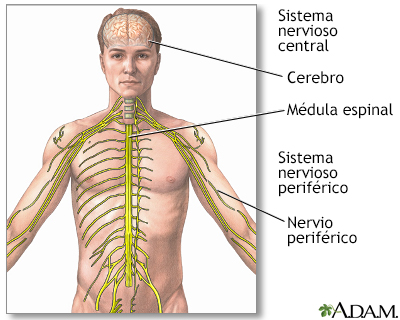 Sistema nervioso ce...
Sistema nervioso ce... Examen de cobre en ...
Examen de cobre en ... Anatomía del hígado
Anatomía del hígado