Distrofia muscular seudohipertrófica; Distrofia muscular de tipo Duchenne DefiniciónEs un trastorno hereditario. Este implica debilidad muscular, la cual empeora rápidamente. CausasLa distrofia muscular de Duchenne es una forma de distrofia muscular que empeora rápidamente. Otras distrofias musculares (incluida la distrofia muscular de Becker) empeoran mucho más lentamente. La distrofia muscular de Duchenne es causada por un gen defectuoso para la distrofina (una proteína en los músculos). Sin embargo, a menudo se presenta en personas con familias sin antecedentes conocidos de esta afección. La afección afecta con mayor frecuencia a los niños debido a la manera en que la enfermedad se hereda. Los hijos de mujeres portadoras de la enfermedad (mujeres con un cromosoma defectuoso, pero que no presentan síntomas) tienen cada uno un 50% de probabilidades de tener la enfermedad y las hijas tienen cada una un 50% de probabilidades de ser portadoras. En ocasiones muy poco frecuentes, una mujer puede ser afectada por la enfermedad. La distrofia muscular de Duchenne se presenta en aproximadamente 1 de cada 3600 varones. Debido a que se trata de un trastorno hereditario, los riesgos incluyen antecedentes familiares de la distrofia muscular de Duchenne. SíntomasLos síntomas frecuentemente aparecen antes de los 6 años de edad. Pueden empezar incluso desde el período de la lactancia. La mayoría de los varones no muestran síntomas en los primeros años de vida. Los síntomas pueden incluir:
Debilidad muscular:
Dificultad progresiva para caminar:
Pruebas y exámenesUn examen completo del sistema nervioso (neurológico), de los pulmones, del corazón y de los músculos puede mostrar:
Los exámenes pueden abarcar:
TratamientoNo existe una cura conocida para la distrofia muscular de Duchenne. El objetivo del tratamiento es controlar los síntomas para optimizar la calidad de vida. Los esteroides pueden disminuir la pérdida de fuerza muscular. El niño puede empezar a tomarlos cuando recibe el diagnóstico o cuando la fuerza muscular comienza a declinar. Otros tratamientos pueden incluir:
Sin embargo, los efectos de estos tratamientos no han sido comprobados. Las células madre y la terapia génica tal vez se usen en el futuro. Las terapias genéticas pueden ser útiles para ciertos pacientes, dependiendo de la causa genética. Cada terapia solo funciona en un pequeño número de pacientes, dependiendo de su cambio (mutación) genético específico. Si bien estos tratamientos han demostrado que aumentan la producción de distrofina, aún no se ha comprobado que tengan beneficios clínicos significativos. Los tratamientos incluyen:
El uso de esteroides y la falta de actividad física pueden conducir a un aumento de peso excesivo. Se fomenta la actividad. La inactividad (como el reposo en cama) puede empeorar la enfermedad muscular. La fisioterapia puede ayudar a mantener la fuerza y el funcionamiento de los músculos. A menudo se necesita terapia del habla. Otros tratamientos pueden incluir:
Se están estudiando varios tratamientos nuevos en ensayos clínicos. Grupos de apoyoEl estrés causado por la enfermedad se puede aliviar uniéndose a un grupo de apoyo, donde los miembros comparten experiencias y problemas en común. La Asociación para la Distrofia Muscular (Muscular Dystrophy Association) es una excelente fuente de información sobre esta enfermedad. Expectativas (pronóstico)La distrofia muscular de Duchenne lleva a una discapacidad que empeora de manera progresiva. A menudo, la muerte ocurre a raíz de trastornos pulmonares, aunque los avances en los cuidados de apoyo han dado como resultado que muchos hombres más tiempo. Posibles complicacionesLas complicaciones pueden incluir:
Cuándo contactar a un profesional médicoContacte a su proveedor de atención médica si:
PrevenciónSe recomienda la asesoría genética si existen antecedentes de la enfermedad en la familia. Los estudios genéticos llevados a cabo durante el embarazo son muy precisos para detectar la distrofia muscular de Duchenne. ReferenciasBharucha-Goebel DX. Muscular dystrophies. In: Kliegman RM, St Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 627. Genetic and Rare Diseases Information Center (GARD). Duchenne muscular dystrophy. rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy. Updated June 2024. Accessed June 11, 2024. Lee BH. The Dystrophinopathies. Continuum (Minneap Minn). 2022;28(6):1678-1697. PMID: 36537975 pubmed.ncbi.nlm.nih.gov/36537975/. Muscular Dystrophy Association website. Duchenne muscular dystrophy. www.mda.org/disease/duchenne-muscular-dystrophy. Accessed March 5, 2024. Selcen D. Muscle diseases. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 389. Warner WC, Sawyer JR. Neuromuscular disorders. In: Azar FM, Beaty JH, eds. Campbell's Operative Orthopaedics. 14th ed. Philadelphia, PA: Elsevier; 2021:chap 35. | ||
| ||
Actualizado : 12/31/2023 Versión en inglés revisada por : Joseph V. Campellone, MD, Department of Neurology, Cooper Medical School at Rowan University, Camden, NJ. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. Editorial update 06/11/2024. Ver referencias La información aquí contenida no debe utilizarse durante ninguna emergencia médica, ni para el diagnóstico o tratamiento de alguna condición médica. Debe consultarse a un médico con licencia para el diagnóstico y tratamiento de todas y cada una de las condiciones médicas. Los enlaces a otros sitios se proporcionan sólo con fines de información, no significa que se les apruebe. No se otorga garantía de ninguna clase, ya sea expresa o implícita, en cuanto a la precisión, confiabilidad, actualidad o exactitud de ninguna de las traducciones hechas por un proveedor de servicios externo de la información aquí contenida en otro idioma. © 1997- A.D.A.M., unidad de negocios de Ebix, Inc. La reproducción o distribución parcial o total de la información aquí contenida está terminantemente prohibida. | ||
| ||


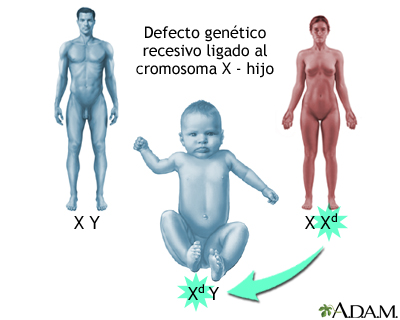 Defectos genéticos ...
Defectos genéticos ...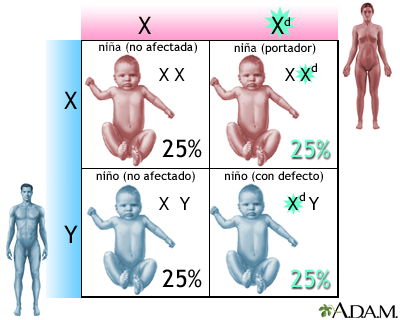 Defectos genéticos ...
Defectos genéticos ...