

Talassemia
Definição
A talassemia é uma doença sanguínea herdada entre os membros de uma família (hereditária) na qual o organismo produz uma forma anormal de hemoglobina. A hemoglobina é a proteína encontrada nos glóbulos vermelhos que carrega o oxigênio. Essa doença resulta em destruição excessiva dos glóbulos vermelhos, causando anemia.
Nomes alternativos
Anemia mediterrânea; Anemia de Cooley; Betatalassemia; Alfatalassemia
Causas
A hemoglobina é formada por duas cadeias de proteínas:
- Alfa
- Beta
A talassemia ocorre quando há um defeito em um gene que ajuda a controlar a produção de uma dessas proteínas.
Existem dois tipos principais de talassemia:
- A alfatalassemia ocorre quando um gene ou genes relacionados à cadeia alfa está ausente ou sofreu uma alteração (mutação).
- A betatalassemia ocorre quando defeitos genéticos similares afetam a produção da cadeia beta.
As alfatalassemias ocorrem mais comumente em pessoas originárias do sudoeste asiático, do Oriente Médio, da China e afrodescendentes.
As betatalassemias ocorrem em indivíduos originários da região do mediterrâneo e, em menor extensão, chineses, demais asiáticos e afrodescendentes.
Existem muitas formas de talassemia. Cada tipo possui muitos subtipos diferentes. Tanto a alfatalassemia quanto a betatalassemia incluem as duas formas abaixo:
- Talassemia maior
- Talassemia menor
É necessário herdar o gene defeituoso de ambos os pais para desenvolver a talassemia maior.
A talassemia menor ocorre se o indivíduo receber o gene defeituoso somente de um dos pais. Indivíduos com essa forma da doença são portadores do distúrbio, mas geralmente não apresentam sintomas.
A betatalassemia maior também é denominada anemia de Cooley.
Os fatores de risco para a talassemia incluem:
- Etnia asiática, chinesa, mediterrânea ou africana
- Histórico familiar da doença
Sintomas
A forma mais grave de alfatalassemia maior causa natimorto (morte do feto durante o parto ou nos últimos estágios da gravidez).
Crianças que nascem com betatalassemia maior (anemia de Cooley) são normais até o nascimento, mas desenvolvem anemia grave durante o primeiro ano de vida.
Outros sintomas podem incluir:
- Deformidades ósseas na face
- Fadiga
- Crescimento retardado
- Falta de ar
- Pele amarela (icterícia)
Indivíduos com a forma menor de alfa e betatalassemia apresentam glóbulos vermelhos pequenos, mas nenhum sintoma.
Sinais e exames
Um exame físico pode revelar aumento do baço.
Uma amostra de sangue é coletada e enviada a um laboratório para ser examinada.
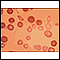
- Os glóbulos vermelhos parecerão pequenos e com formato anormal quando observados ao microscópio.
- Um hemograma completo revela anemia.
- Um exame denominado eletroforese de hemoglobina identifica a presença de uma forma anormal de hemoglobina.
- Um exame denominado análise mutacional pode ajudar a detectar a alfatalassemia.
Tratamento
O tratamento para talassemia maior geralmente envolve transfusões sanguíneas regulares e suplementos de folato.
Caso receba transfusões de sangue, o indivíduo não deve tomar suplementos de ferro. Isso poderia acarretar um aumento grande da quantidade de ferro no organismo, o que pode ser prejudicial.
Indivíduos que recebem um número significativo de transfusões sanguíneas necessitam de um tratamento denominado terapia de quelação, para eliminar o excesso de ferro no organismo.
Um transplante de medula óssea pode ajudar a tratar a doença em alguns pacientes, especialmente em crianças.
Expectativas (prognóstico)
A talassemia grave pode causar morte prematura devido à insuficiência cardíaca, geralmente os entre 20 e 30 anos de idade. Transfusões sanguíneas regulares e terapia para eliminar o ferro do organismo ajudam a melhorar o prognóstico.
Formas menos graves de talassemia, em geral, não diminuem a expectativa de vida.
Aconselhamento genético e exames de pré-natal podem ajudar indivíduos com histórico familiar dessa condição que estejam planejando ter filhos.
Complicações
A talassemia maior não tratada causa insuficiência cardíaca e problemas no fígado, além de deixar o indivíduo mais suscetível a desenvolver infecção.
Transfusões sanguíneas podem ajudar a controlar alguns sintomas. Entretanto, elas podem resultar em uma quantidade excessiva de ferro.
Quando contatar um profissional de saúde
Entre em contato com o seu médico se:
- Você ou seu filho apresentar sintomas de talassemia
- Estiver recebendo tratamento para a doença e novos sintomas se desenvolverem
Referências
Cappellini MD. The thalassemias. In: Goldman L, Schafer AI, eds. Goldman's Cecil Medicine. 25th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap 162.
DeBaun MR, Frei-Jones MJ, Vichinsky EP. Hemoglobinopatiies. In: Kliegman RM, Stanton BF, St Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20th ed. Philadelphia, PA: Elsevier; 2016:chap 462.
Giardina PJ, Rivella S. Thalassemia syndromes. In: Hoffman R, Benz EJ Jr, Silberstein LE, Heslop HE, Weitz JI, Anastasi J, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:chap 38.
Revisão feita por: Todd Gersten, MD, Hematology/Oncology, Florida Cancer Specialists & Research Institute, Wellington, FL. Review provided by VeriMed Healthcare Network. Also reviewed by David Zieve, MD, MHA, Isla Ogilvie, PhD, and the A.D.A.M. Editorial team.
 Todos os direitos reservados
Todos os direitos reservados