Deficiencia de glucosilceramidasa; Deficiencia de glucocerebrosidasa; Enfermedad por depósito lisosomal - Gaucher DefiniciónEs un trastorno genético poco común en el cual una persona carece de una enzima llamada glucocerebrosidasa (GBA). CausasLa enfermedad de Gaucher es poco común en la población general. Las personas con ascendencia judía oriunda de Europa Central y Oriental (asquenazíes) son más susceptibles a presentarla. Se trata de una enfermedad autosómica recesiva. Esto significa que tanto la madre como el padre tendrían que transmitirle una copia de la variante del gen de la enfermedad a su hijo para que presente la enfermedad. Un padre que porta una copia de la variante del gen, pero no presenta la enfermedad se denomina portador silencioso. La falta de GBA hace que se acumulen sustancias dañinas en el hígado, el bazo, los huesos y la médula ósea. Estas sustancias impiden que células y órganos funcionen apropiadamente. Existen tres subtipos de la enfermedad de Gaucher:
SíntomasEl sangrado debido al bajo conteo de plaquetas es el síntoma más común observado en la enfermedad de Gaucher. Otros síntomas pueden incluir:
Pruebas y exámenesSu proveedor de atención médica le realizará un examen físico y le preguntará sobre sus síntomas. Se pueden realizar los siguientes exámenes:
TratamientoLa enfermedad de Gaucher no tiene cura. Sin embargo, los tratamientos pueden ayudar a regular y aliviar los síntomas. Se pueden suministrar medicamentos para:
Otros tratamientos incluyen:
Grupos de apoyoEstos grupos pueden proporcionar más información acerca de la enfermedad de Gaucher:
Expectativas (pronóstico)La recuperación de una persona depende del subtipo de la enfermedad. La forma infantil de esta enfermedad (tipo 2) puede conducir a la muerte temprana. La mayoría de los niños afectados muere antes de los 5 años de edad. Los adultos con el tipo 1 de la enfermedad Gaucher pueden tener la expectativa de una vida normal con la terapia de reemplazo enzimático. Posibles complicacionesLas complicaciones de la enfermedad de Gaucher pueden incluir:
PrevenciónSe recomienda la asesoría genética para los futuros padres con antecedentes familiares de la enfermedad de Gaucher. Con las pruebas, se puede determinar si ambos padres son portadores de una copia de la variante del gen que por lo tanto podría transmitir la enfermedad. También es posible determinar en un examen prenatal si un bebé en el vientre materno tiene esta enfermedad. ReferenciasKliegman RM, St. Geme JW, Blum NJ, et al. Defects in metabolism of lipids. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 106. Krasnewich DM, Sidransky E. Lysosomal storage diseases. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 192. Turnpenny PD, Ellard S, Cleaver R. Inborn errors of metabolism. In: Turnpenny PD, Ellard S, Cleaver R, eds. Emery's Elements of Medical Genetics and Genomics. 16th ed. Philadelphia, PA: Elsevier; 2022:chap 18. | ||
| ||
Actualizado : 11/6/2024 Versión en inglés revisada por : Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. Ver referencias La información aquí contenida no debe utilizarse durante ninguna emergencia médica, ni para el diagnóstico o tratamiento de alguna condición médica. Debe consultarse a un médico con licencia para el diagnóstico y tratamiento de todas y cada una de las condiciones médicas. Los enlaces a otros sitios se proporcionan sólo con fines de información, no significa que se les apruebe. No se otorga garantía de ninguna clase, ya sea expresa o implícita, en cuanto a la precisión, confiabilidad, actualidad o exactitud de ninguna de las traducciones hechas por un proveedor de servicios externo de la información aquí contenida en otro idioma. © 1997- A.D.A.M., unidad de negocios de Ebix, Inc. La reproducción o distribución parcial o total de la información aquí contenida está terminantemente prohibida. | ||
| ||


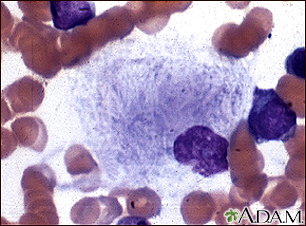 Microfotografía de ...
Microfotografía de ...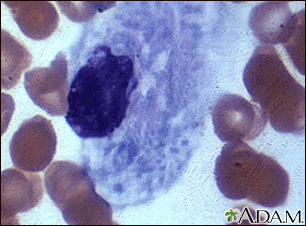 Microfotografía 2 d...
Microfotografía 2 d...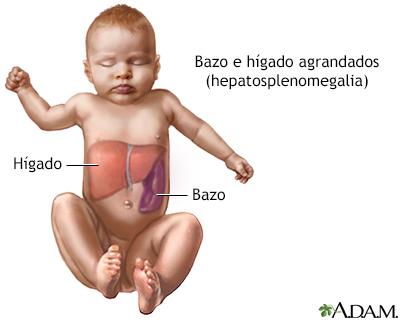 Hepatoesplenomegali...
Hepatoesplenomegali...