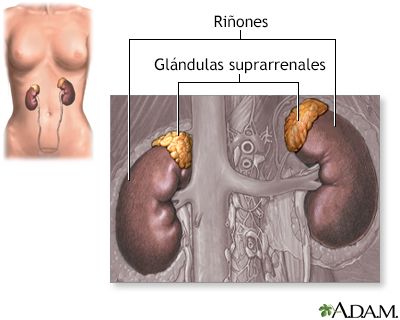
Síndrome adrenogenital; Déficil de 21 hidroxilasa; HSC DefiniciónEs el nombre dado a un grupo de trastornos hereditarios de las glándulas suprarrenales. Hereditario significa que los rasgos se transmiten de padres a hijos. CausasUsted tiene 2 glándulas suprarrenales. Son dos órganos del tamaño de una nuez de nogal que están colocadas sobre cada uno de los riñones. Las glándulas suprarrenales producen las siguientes hormonas que son esenciales para la vida.
A la mayoría de las personas con hiperplasia suprarrenal congénita (HSC) les hace falta una enzima llamada 21-hidroxilasa. Las glándulas suprarrenales necesitan esta enzima para producir suficiente cortisol y aldosterona. Debido a los bajos niveles de cortisol, el cuerpo estimula a las glándulas suprarrenales, que luego producen más andrógenos (Este es el resultado de un desbalance de estas hormonas). Existen dos tipos principales de HSC que conforman la mayoría de los casos: HSC clásica y HSC no clásica. HSC clásica es poco frecuente y más grave. Por lo regular se detecta al nacer o durante la primera infancia. El cuerpo produce muy poca aldosterona y cortisol y demasiados andrógenos. Existen dos subtipos de HSC:
HSC no clásica es la forma más leve y común. El cuerpo produce suficiente aldosterona y cortisol, pero no muchos andrógenos. Con frecuencia no se diagnostica hasta la etapa tardía de la niñez o la adultez. La persona puede tener síntomas leves o no presentar síntomas. SíntomasLos síntomas varían, dependiendo del tipo de HSC que alguien tenga y de su edad cuando se diagnostica el trastorno.
Los niños con HSC clásica perdedora de sal, a menudo presentan síntomas suprarrenales más graves al cabo de 2 o 3 semanas después del nacimiento. Estos pueden incluir:
Las mujeres con HSC no clásica generalmente tendrán órganos reproductores femeninos normales (ovarios, útero y trompas de Falopio). También pueden tener los siguientes cambios:
Los hombres con HSC no clásica a menudo parecen normales al nacer. Sin embargo, puede parecer que llegan temprano a la pubertad. Los síntomas pueden incluir:
Tanto los hombres como las mujeres crecerán rápidamente durante la niñez, pero serán mucho más bajos de lo normal como adultos. Pruebas y exámenesSi usted tiene antecedentes familiares de HSC, es posible que quiera hablar con su proveedor de atención médica acerca de realizarse pruebas prenatales para buscar el trastorno en su bebé nonato: Al nacer, a su hijo se le hará la prueba de HSC como parte de las pruebas de detección neonatales. Se hace una punción del talón para extraer sangre (como parte de las pruebas de detección de rutina que se realizan a recién nacidos). Sin embargo, esta solo puede detectar HSC clásico. Si una persona tiene síntomas de cualquier tipo de HSC, el proveedor realizará un examen y ordenará ciertas pruebas. Los análisis de sangre comunes incluyen: Las pruebas genéticas pueden ayudar a diagnosticar o confirmar el trastorno, pero se necesitan en muy pocos casos. TratamientoEl objetivo del tratamiento es devolver los niveles hormonales a lo normal o cerca de lo normal. El tratamiento puede incluir:
Los medicamentos deben tomarse diariamente. Las personas pueden necesitar dosis adicionales de medicamentos en períodos de estrés, como una enfermedad grave o una cirugía. El monitoreo vitalicio es necesario para asegurar que se mantenga un nivel hormonal adecuado. Los esteroides (como la hidrocortisona y la fludrocortisona) empleados para tratar la HSC por lo general no causan efectos secundarios, como obesidad o huesos débiles, ya que las dosis reponen las hormonas que el cuerpo no puede producir. Los esteroides no se pueden suspender de manera súbita, ya que hacer esto puede provocar una crisis suprarrenal. Las personas con HSC no clásica posiblemente solo necesitarán dosis bajas de medicamentos o no los necesiten en absoluto. Un equipo de profesionales de atención médica con experiencia en HSC trabajará junto para tratar a su hijo y apoyar a la familia. Este equipo puede incluir neonatólogos, genetistas, endocrinólogos y psiquiatras o trabajadores sociales. Las bebés con genitalia ambigua pueden necesitar cirugía para mejorar su función y crear una apariencia más típica de mujer. Muchos expertos en salud sugieren esperar hasta que el niño sea mayor para participar de esta decisión, salvo que la cirugía sea necesaria para la salud del bebé. Hable con los proveedores de atención médica de su hijo acerca de qué es lo mejor. Trabajar con un profesional de la salud mental es una parte fundamental de un plan de tratamiento para niños con HSC y sus familias. Grupos de apoyoPuede obtener más información y apoyo para las familias con HSC en sus familias en:
Expectativas (pronóstico)La mayoría de las personas con este trastorno deben tomar medicamentos de por vida. En su mayoría gozan de buena salud. Sin embargo, pueden ser más bajas que los adultos normales, incluso con tratamiento. En algunos casos, la HSC puede afectar la fertilidad. Posibles complicacionesUna crisis suprarrenal es una complicación grave de la HSC clásica. PrevenciónDebido a que esta es una afección genética, no hay manera de prevenirla. Sin embargo, los padres con antecedentes familiares de HSC (de cualquier tipo) o un niño que padezca esta afección deben pensar en solicitar asesoría genética. ReferenciasDonohoue PA. Disorders of sex development. In: Kliegman RM, St Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 606. Escobar O, Gurtunca N, Viswanathan P, Witchel SF. Pediatric endocrinology. In: Zitelli BJ, McIntire SC, Nowalk AJ, Garrison J, eds. Zitelli and Davis' Atlas of Pediatric Physical Diagnosis. 8th ed. Philadelphia, PA: Elsevier; 2023:chap 9. Eunice Kennedy Shriver National Institute of Child Health and Human Development website. Congenital adrenal hyperplasia (CAH). www.nichd.nih.gov/health/topics/cah. Updated 5/17/2021. Accessed January 11, 2024. Newell-Price, JDC, Auchus RJ. The adrenal cortex. In: Melmed S, Auchus, RJ, Goldfine AB, Koenig RJ, Rosen CJ, eds. Williams Textbook of Endocrinology. 14th ed. Philadelphia, PA: Elsevier; 2020:chap 15. | ||
| ||
Actualizado : 3/12/2024 Versión en inglés revisada por : Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. Ver referencias La información aquí contenida no debe utilizarse durante ninguna emergencia médica, ni para el diagnóstico o tratamiento de alguna condición médica. Debe consultarse a un médico con licencia para el diagnóstico y tratamiento de todas y cada una de las condiciones médicas. Los enlaces a otros sitios se proporcionan sólo con fines de información, no significa que se les apruebe. No se otorga garantía de ninguna clase, ya sea expresa o implícita, en cuanto a la precisión, confiabilidad, actualidad o exactitud de ninguna de las traducciones hechas por un proveedor de servicios externo de la información aquí contenida en otro idioma. © 1997- A.D.A.M., unidad de negocios de Ebix, Inc. La reproducción o distribución parcial o total de la información aquí contenida está terminantemente prohibida. | ||
| ||


 Glándulas suprarren...
Glándulas suprarren...