Deficiencia de galactosa-1-fosfatouridil transferasa; Deficiencia de galactocinasa; Deficiencia de galactosa-6-fosfato epimerasa; GALT; GALK; GALE; Galactosemia por deficiencia de epimerasa; Deficiencia de GALE; Galactosemia tipo III; UDP - galactosa-4; Variante de Duarte DefiniciónEs una afección en la cual el cuerpo no puede utilizar (metabolizar) el azúcar simple galactosa. CausasLa galactosemia es un trastorno hereditario. Esto quiere decir que se transmite de padres a hijos. Si ambos padres portan una copia defectuosa del gen que causa esta enfermedad, cada uno de sus hijos tiene un 25% (1 en 4) de probabilidades de resultar afectado por ella. Esto se denomina herencia autosómica recesiva. Existen 3 formas de la enfermedad:
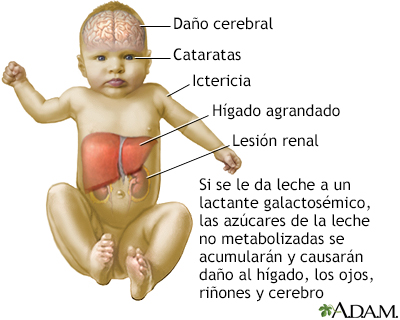
Las personas con galactosemia son incapaces de descomponer completamente el azúcar simple galactosa. La galactosa compone la mitad de la lactosa, el azúcar que se encuentra en la leche. Si a un bebé con galactosemia se le da leche, los derivados de la galactosa se acumulan en el organismo del bebé. Estas sustancias dañan el hígado, el cerebro, los riñones y los ojos. Las personas con galactosemia no pueden tolerar ninguna forma de leche (ni humana ni animal). Deben ser cuidadosos al consumir otros alimentos que contengan galactosa. SíntomasLos bebés con galactosemia pueden mostrar síntomas en los primeros días de vida si consumen leche de fórmula o leche materna que contengan lactosa. Estos pueden desarrollar una infección grave en la sangre con la bacteria E. coli. Los síntomas de la galactosemia son:
Pruebas y exámenesLos exámenes para detectar la galactosemia incluyen:
En muchos estados, las pruebas de detección en recién nacidos evalúan esta afección. Los resultados de los exámenes pueden mostrar:
TratamientoLas personas que padezcan esta afección deben evitar de por vida todos los tipos de leche, los productos que contengan leche (incluso la leche en polvo) y otros alimentos que contengan galactosa. Lea las etiquetas de los alimentos para asegurarse de que usted o un hijo que presente la afección no estén consumiendo alimentos que contengan galactosa. A los bebés se les puede dar de comer:
Se recomiendan ciertos suplementos de calcio. Grupos de apoyoSe puede encontrar más información y apoyo para las personas con galactosemia y sus familias en: Galactosemia Foundation -- www.galactosemia.org Expectativas (pronóstico)Las personas que reciben un diagnóstico temprano y que evitan estrictamente los productos lácteos y otros alimentos que contienen lactosa pueden llevar una vida relativamente normal. Sin embargo, se puede presentar un leve deterioro mental, incluso en personas que evitan la galactosa. Posibles complicacionesSe pueden presentar las siguientes complicaciones:
Cuándo contactar a un profesional médicoComuníquese con proveedor de atención médica si:
PrevenciónConocer los antecedentes familiares es de gran ayuda. Si usted tiene antecedentes familiares de galactosemia y desea tener hijos, la asesoría genética le ayudará a tomar una decisión sobre el embarazo y las pruebas prenatales. Una vez que se hace el diagnóstico de galactosemia, se recomienda la asesoría genética para otros miembros de la familia. Muchos estados (en los Estados Unidos) hacen exámenes de detección de galactosemia en recién nacidos. Si los exámenes del recién nacido indican la posible presencia de esta enfermedad, deben comunicarse con el proveedor del niño de inmediato para obtener consejos sobre cómo administrar productos lácteos a sus bebés. También deberían solicitarle al proveedor exámenes de sangre que se puedan hacer para confirmar un diagnóstico de galactosemia. ReferenciasBerry GT. Classic galactosemia and clinical variant galactosemia. 2000 Feb 4 [updated 2021 Mar 11]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews [Internet]. Seattle, WA: University of Washington. PMID: 20301691 pubmed.ncbi.nlm.nih.gov/20301691/. Bonnardeaux A, Bichet DG. Inherited disorders of the renal tubule. In: Yu ASL, Chertow GM, Luyckx VA, Marsden PA, Skorecki K, Taal MW, eds. Brenner and Rector's The Kidney. 11th ed. Philadelphia, PA: Elsevier; 2020:chap 44. Hijazi G, Kishnani PS. Defects in metabolism of carbohydrates. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 107. Pearl PL, DiBacco ML, Gibson KM. Inborn errors of metabolism and the nervous system. In: Jankovic J, Mazziotta JC, Pomeroy SL, Newman NJ, eds. Bradley and Daroff's Neurology in Clinical Practice. 8th ed. Philadelphia, PA: Elsevier; 2022:chap 91. | ||
| ||
Actualizado : 4/8/2025 Versión en inglés revisada por : Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. Ver referencias La información aquí contenida no debe utilizarse durante ninguna emergencia médica, ni para el diagnóstico o tratamiento de alguna condición médica. Debe consultarse a un médico con licencia para el diagnóstico y tratamiento de todas y cada una de las condiciones médicas. Los enlaces a otros sitios se proporcionan sólo con fines de información, no significa que se les apruebe. No se otorga garantía de ninguna clase, ya sea expresa o implícita, en cuanto a la precisión, confiabilidad, actualidad o exactitud de ninguna de las traducciones hechas por un proveedor de servicios externo de la información aquí contenida en otro idioma. © 1997- A.D.A.M., unidad de negocios de Ebix, Inc. La reproducción o distribución parcial o total de la información aquí contenida está terminantemente prohibida. | ||
| ||


 Galactosemia
Galactosemia