




Fibrose cística
Definição
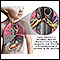
A fibrose cística é uma doença hereditária que causa o acúmulo de muco denso e pegajoso nos pulmões, no trato digestivo e outras áreas do corpo. É uma das doenças pulmonares crônicas mais comuns em crianças e adultos jovens, sendo um transtorno potencialmente fatal.
Nomes alternativos
Mucoviscidose
Causas
A fibrose cística é uma doença hereditária, causada por um gene defeituoso que faz com que o corpo produza secreção (muco) anormalmente espessa e pegajosa. O muco se acumula nas passagens respiratórias dos pulmões e no pâncreas.
O acúmulo do muco espesso resulta em infecções pulmonares que colocam a vida em risco, além de problemas digestivos graves. A doença também pode afetar as glândulas sudoríparas e o sistema reprodutivo masculino.
Muitas pessoas têm o gene defeituoso para fibrose cística, mas não têm nenhum sintoma. Isto acontece porque a pessoa precisa herdar dois genes defeituosos, um de cada progenitor, para que a doença se manifeste. A fibrose cística é mais comum entre descendentes de europeus nórdicos ou centrais.
A maioria das crianças com fibrose cística é diagnosticada até 2 anos de idade. Um número menor, no entanto, não é diagnosticado até os 18 anos ou mais. Estes pacientes geralmente têm uma forma mais branda da doença.
Sintomas
Os sintomas em recém-nascidos podem incluir:
- Crescimento retardado
- Dificuldade para ganhar peso normalmente durante a infância
- Nenhuma evacuação nas primeiras 24 a 48 horas de vida
- Pele salgada
Os sintomas relacionados ao funcionamento do intestino podem incluir:
- Dor no abdome devido à constipação grave
- Aumento de gases, inchaço ou abdome que parece inchado (dilatado)
- Náusea e perda de apetite
- Fezes pálidas ou cor de massa de vidraceiro, odor fétido, com muco ou flutuantes
- Perda de peso
Os sintomas relativos aos pulmões e seios paranasais podem incluir:
- Tosse ou aumento de muco nos seios paranasais ou pulmões
- Fadiga
- Congestão nasal causada por pólipos nasais
- Episódios recorrentes de pneumonia (sintomas de pneumonia em uma pessoa com fibrose cística incluem febre, aumento da tosse e da dificuldade para respiratória, aumento do muco e perda de apetite)
- Dor ou pressão nos seios paranasais causada por infecção ou pólipos
Sintomas que podem ser observados em pessoas de idade mais avançada:
- Infertilidade (em homens)
- Inflamação repetida do pâncreas (pancreatite)
- Sintomas respiratórios
- Baqueteamento (hipocratismo) digital
Sinais e testes
Um teste de sangue está disponível para ajudar a detectar a fibrose cística. O teste procura por variações em um gene conhecido que causa a doença. Outros testes usados para diagnosticar a fibrose cística incluem:
- O teste do tripsinogênio imunorreativo (IRT) é um teste padrão de triagem em recém-nascidos. Um alto nível de IRT sugere possível fibrose cística e requer investigação adicional.
- O teste de cloro no suor é o teste de diagnóstico padrão para fibrose cística. Um alto nível de sal no suor do paciente é sinal da doença.
Outros testes que identificam problemas que podem estar relacionados à fibrose cística incluem:
- Raio-X ou tomografia computadorizada do tórax
- Teste de gordura nas fezes
- Testes de função pulmonar
- Medição da função pancreática
- Teste de estimulação da secretina
- Tripsina e quimotripsina nas fezes
- Estudo contrastado do trato gastrointestinal superior e do intestino delgado
Tratamento
Um diagnóstico precoce de fibrose cística e um plano de tratamento abrangente podem melhorar a sobrevivência e a qualidade de vida. O acompanhamento e monitoramento são muito importantes. Se possível, os pacientes devem ser atendidos em um centro especializado em fibrose cística. Quando as crianças atingirem a idade adulta, elas devem ser transferidas para um centro especializado em fibrose cística para adultos.
O tratamento para problemas pulmonares inclui:
- Antibióticos para prevenir e tratar de infecções nos pulmões e nos seios paranasais. Eles podem ser tomados por via oral ou aplicados nas veias ou inalados. As pessoas com fibrose cística podem tomar antibióticos somente quando necessário ou o tempo todo. As doses geralmente são maiores do que o normal. Siga as recomendações do seu médico.
- Outros medicamentos inalados que deixam o muco menos espesso e facilitam sua eliminação através da tosse (expectoração) são terapia com enzima DNAse recombinante e soluções salinas hipertônicas.
- Vacina contra influenza e vacina antipneumocócica polissacarídica (PPV) anualmente (converse com o seu médico).
- O transplante de pulmão é uma opção em alguns casos.
- A oxigenoterapia pode ser necessária à medida que a doença pulmonar piora.
Problemas pulmonares também são tratados com outras terapias para afinar o muco e facilitar a expectoração. Tais métodos incluem:
- Atividade física ou exercícios de respiração profunda
- Aparelhos usados durante o dia que ajudam a eliminar o excesso de muco das vias aéreas
- Percussão torácica manual (fisioterapia) na qual um membro da família ou um fisioterapeuta dá pancadas leves no tórax, nas costas e na área sob os braços do paciente com fibrose cística
Tratamentos de problemas intestinais e nutricionais podem incluir:
- Uma dieta especial rica em proteínas e calorias para crianças maiores e adultos
- Enzimas pancreáticas para ajudar a absorver gorduras e proteína, tomadas a cada refeição
- Suplementos vitamínicos, especialmente vitaminas A, D, E e K
- Seu médico pode sugerir outros tratamentos se você tiver fezes muito duras
Ivacaftor é um medicamento que trata certos tipos de fibrose cística, melhorando a função de um dos genes defeituosos que causa a doença. Como resultado, há menos acumulação de muco espessado nos pulmões. Outros sintomas também podem apresentar melhora. Este medicamento foi aprovado pela agência reguladora norte-americana FDA recentemente, mas ainda não possui registro na Anvisa, não sendo comercializado no Brasil.
Cuidados e monitoramento em casa devem incluir:
- Evitar o fumo, a poeira, a sujeira, as fumaças, os produtos químicos domésticos, a fumaça de lareira e o mofo ou bolor.
- Beber líquidos em abundância, principalmente bebês e crianças vivendo em clima quente, quando houver diarreia ou fezes moles ou durante atividade física adicional.
- Exercícios duas ou três vezes por semana. Natação, corrida e ciclismo são boas opções.
- Limpar ou retirar muco ou secreções das vias respiratórias. Isso deve ser feito de uma a quatro vezes por dia. Pacientes, familiares e profissionais de saúde devem aprender sobre como fazer percussão torácica e drenagem postural para ajudar a manter as vias respiratórias limpas.
Grupos de apoio
Você pode aliviar o estresse da doença frequentando um grupo de apoio à fibrose cística. Compartilhar com outras pessoas que têm experiências e problemas em comum pode ajudar você e a sua família a não se sentirem sozinhos.
Expectativa (prognóstico)
A maioria das crianças com fibrose cística permanece razoavelmente saudável até atingir a idade adulta. Elas podem participar da maioria das atividades e devem frequentar a escola. Muitos adultos jovens com fibrose cística terminam a faculdade ou encontram emprego.
A doença pulmonar piora ao ponto de a pessoa ficar incapacitada. Hoje, a expectativa de vida média para pessoas com fibrose cística que vivem até a idade adulta é de aproximadamente 37 anos.
O óbito é geralmente causado por complicações pulmonares.
Complicações
A complicação mais comum é a infecção respiratória crônica.
Outras complicações incluem:
- Problemas intestinais, como cálculos biliares, obstrução intestinal e prolapso retal
- Expectoração de sangue
- Insuficiência respiratória crônica
- Diabetes
- Infertilidade
- Doença hepática ou insuficiência hepática, pancreatite, cirrose biliar
- Desnutrição
- Pólipos nasais e sinusite
- Osteoporose e artrite
- Pneumonia recorrente
- Pneumotórax
- Insuficiência cardíaca no lado direito (cor pulmonale)
Quando contatar um profissional de saúde
Entre em contato com o seu médico se o bebê ou a criança tiver sintomas de fibrose cística.
Ligue para o seu médico se você ou seu filho tiver:
- Febre, aumento de tosse, alterações na saliva ou sangue na saliva, perda de apetite ou outros sinais de pneumonia
- Aumento na perda de peso
- Evacuações mais frequentes ou fezes com odor fétido, ou com mais muco
- Abdome inchado ou aumento no intumescimento
Ligue para o seu médico se uma pessoa com fibrose cística desenvolver sintomas novos ou se os sintomas piorarem, especialmente dificuldade respiratória grave ou expectoração de sangue.
Prevenção
Não existe nenhuma maneira de prevenir a fibrose cística. A triagem das pessoas com histórico familiar da doença pode detectar o gene da fibrose cística em portadores.
Referências
Accurso FJ. Cystic fibrosis. In: Goldman L, Schafer AI, eds. Goldman's Cecil Medicine. 25th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap 89.
Cutting GR. Cystic fibrosis. In: Rimoin DL, Pyeritz RE, Korf BR, eds. Emery and Rimoin's Principles and Practice of Medical Genetics. 6th ed. Philadelphia, PA: Elsevier; 2013:chap 58.
Egan ME, Green DM, Voynow JA. Cystic fibrosis. In: Kliegman RM, Stanton BF, St Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20th ed. Philadelphia, PA: Elsevier; 2016:chap 403.
Revisão feita por: Neil K. Kaneshiro, MD, MHA, Clinical Professor of Pediatrics, University of Washington School of Medicine, Seattle, WA. Also reviewed by David Zieve, MD, MHA, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
 Todos os direitos reservados
Todos os direitos reservados