CF DefinitionCystic fibrosis is a disease that causes thick, sticky mucus to build up in the lungs, digestive tract, and other areas of the body. It is one of the most common chronic lung diseases in children and young adults. It is a life-threatening disorder. CausesCystic fibrosis (CF) is a disease that is passed down through families. It is caused by a defective gene that makes the body produce abnormally thick and sticky fluid, called mucus. This mucus builds up in the breathing passages of the lungs and in the pancreas. The buildup of mucus results in life-threatening lung infections and serious digestion problems. The disease may also affect the sweat glands and a man's reproductive system. Many people carry a CF gene, but do not have symptoms. This is because for a person to get CF, they must inherit 2 defective genes, 1 from each parent. Some Americans have the CF gene. It is more common among those of northern or central European descent. Most children with CF are diagnosed by age 2, especially as newborn screening is performed across the United States. For a small number, the disease is not detected until age 18 or older. These children often have a milder form of the disease. SymptomsSymptoms in newborns may include:
Symptoms related to bowel function may include:
Symptoms related to the lungs and sinuses may include:
Symptoms that may be noticed later in life:
Exams and TestsA blood test is done to help detect CF. The test looks for changes in the CF gene. Other tests used to diagnose CF include:
Other tests that identify problems that can be related to CF include:
TreatmentAn early diagnosis of CF and treatment plan can improve both survival and quality of life. Follow-up and monitoring are very important. When possible, care should be received at a cystic fibrosis specialty clinic. When children reach adulthood, they should transfer to a cystic fibrosis specialty center for adults. Treatment for lung problems includes:
Lung problems are also treated with therapies to thin the mucus. This makes it easier to cough the mucus out of the lungs. These methods include:
Treatment for bowel and nutritional problems may include:
Ivacaftor, lumacaftor, tezacaftor, and elexacaftor are medicines that treat certain types of CF.
Care and monitoring at home should include:
Support GroupsYou can ease the stress of illness by joining a cystic fibrosis support group. Sharing with others who have common experiences and problems can help your family to not feel alone. Outlook (Prognosis)Most children with CF stay in good health until they reach adulthood. They are able to take part in most activities and attend school. Many young adults with CF finish college or find jobs. Lung disease eventually worsens to the point where the person is disabled. Today, the average life span for people with CF who live to adulthood is about 44 years. Death is most often caused by lung complications. Possible ComplicationsThe most common complication is chronic respiratory infection. Other complications include:
When to Contact a Medical ProfessionalContact your provider if an infant or child has symptoms of CF, and experiences:
Contact your provider if a person with CF develops new symptoms or if symptoms get worse, particularly severe breathing difficulty or coughing up blood. PreventionCF cannot be prevented. Screening those with a family history of the disease may detect the CF gene in many carriers. ReferencesEagan ME, Schechter MS, Voynow JA. Cystic fibrosis. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 432. Grasemann H. Cystic fibrosis. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 77. Solomon GM, Hoover W, Sorscher EJ, Rowe SM. Cystic fibrosis: diagnosis and management. In: Broaddus VC, Ernst JD, King TE, et al, eds. Murray and Nadel's Textbook of Respiratory Medicine. 7th ed. Philadelphia, PA: Elsevier; 2022:chap 68. Phan H, Daines CL. Cystic fibrosis. In: Kellerman RD, Rakel DP, Heidelbaugh JJ, Lee EM, eds. Conn's Current Therapy 2024. Philadelphia, PA: Elsevier; 2024:923-928. | ||
| ||
Review Date: 2/17/2024 Reviewed By: Charles I. Schwartz, MD, FAAP, Clinical Assistant Professor of Pediatrics, Perelman School of Medicine at the University of Pennsylvania, General Pediatrician at PennCare for Kids, Phoenixville, PA. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. View References The information provided herein should not be used during any medical emergency or for the diagnosis or treatment of any medical condition. A licensed medical professional should be consulted for diagnosis and treatment of any and all medical conditions. Links to other sites are provided for information only -- they do not constitute endorsements of those other sites. No warranty of any kind, either expressed or implied, is made as to the accuracy, reliability, timeliness, or correctness of any translations made by a third-party service of the information provided herein into any other language. © 1997- A.D.A.M., a business unit of Ebix, Inc. Any duplication or distribution of the information contained herein is strictly prohibited. | ||
| ||


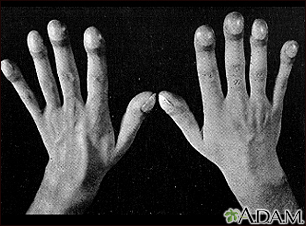 Clubbing
Clubbing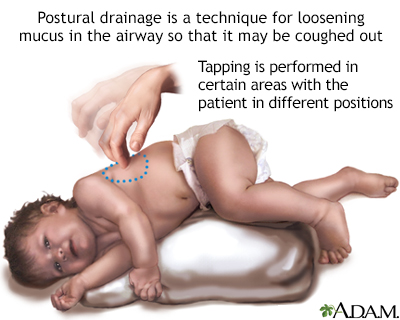 Postural drainage
Postural drainage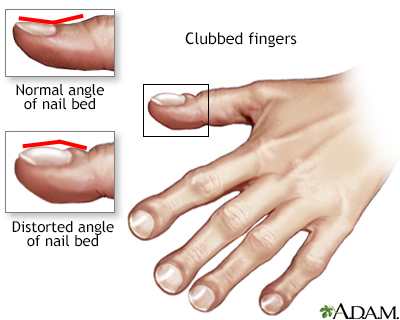 Clubbed fingers
Clubbed fingers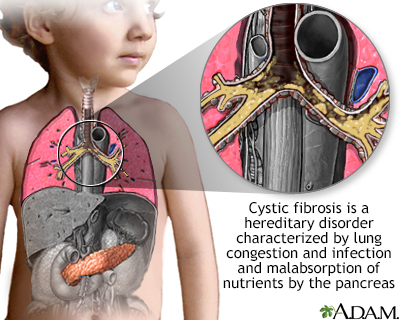 Cystic fibrosis
Cystic fibrosis